Usage¶
Importing promoe into your own project:¶
import promoe
promoe’s commandline interface:¶
Promoe offers three not necessarily distinct entry points:
____ ____ ___ __ __ ___ _____
| _ \| _ \ / _ \| \/ |/ _ \| ____|
| |_) | |_) | | | | |\/| | | | | _|
| __/| _ <| |_| | | | | |_| | |___
|_| |_| \_ \___/|_| |_|\___/|_____|
Usage: promoe [OPTIONS] COMMAND [ARGS]…
- Options:
-v, --verbose Verbose output (print debug statements) --help Show this message and exit.
Commands:
protonize
protonize-selected
clean-hydrogens
The help menus of the respective commands can be requested via:
promoe <command> --help
e.g. promoe protonize --help
promoe protonize¶
Help menu:
| _ \| _ \ / _ \| \/ |/ _ \| ____|
| |_) | |_) | | | | |\/| | | | | _|
| __/| _ <| |_| | | | | |_| | |___
|_| |_| \_ \___/|_| |_|\___/|_____|
Usage: promoe protonize [OPTIONS]
- Options:
--pdbs TEXT pdb identifier to protonize -> binding site will be extracted automatically [required] --keep_hydrogens Specify if all hydrogens after protonization should be kept. --remove_hydrogens Specify if hydrogens that are not within a specific distance should be deleted. --distance INTEGER The maximal distance of the ligand to the surrounding water atoms after protonization, that should be kept. Default is 4. Hence, all water atoms within distance 4 to the ligand are not removed! --help Show this message and exit
Multiple pdbs can be specified by repeating the –pdb parameter.
The workflow is as follows:
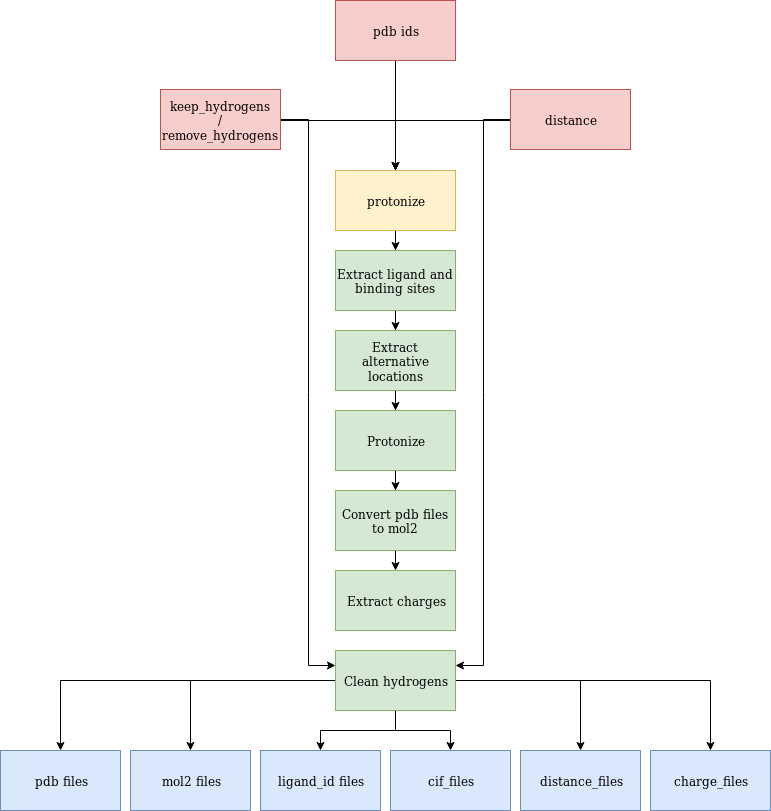
The results include several folders separating the generated results.
The pdb, as well as mol2 files for the full protein, the binding sites, as well as the ligands can be found in the pdb_files and mol2_files folders.
Additionally, the protonated binding sites are contained in the mol2 folder.
All distances from the atoms to the ligand that are below the specified distance are in the distances_files folder.
Moreover, the calculated charges for all residues are contained in the charge_files folder.
promoe protonize-selected¶
Help menu:
____ ____ ___ __ __ ___ _____
| _ \| _ \ / _ \| \/ |/ _ \| ____|
| |_) | |_) | | | | |\/| | | | | _|
| __/| _ <| |_| | | | | |_| | |___
|_| |_| \_ \___/|_| |_|\___/|_____|
Usage: promoe protonize-selected [OPTIONS]
- Options:
--pdb TEXT Path to a pdb file [required] --atoms TEXT A list of atoms in the following format: –atoms ‘[‘GLN’, ‘144’, ‘N’], [‘LEU’, ‘145’, ‘N’]’ [required] --chain TEXT A chain specifier such as ‘A’ or ‘B’ or ‘all’ --keep_hydrogens Specify if all hydrogens after protonization should be kept. --remove_hydrogens Specify if hydrogens that are not within a specific distance should be deleted. --distance INTEGER The maximal distance of the ligand to the surrounding water atoms after protonization, that should be kept. Default is 4. Hence, all water atoms within distance 4 to the ligand are not removed! --help Show this message and exit.
Please ensure, that the atoms are specified in the following format: ‘[‘GLN’, ‘144’, ‘N’], [‘LEU’, ‘145’, ‘N’]’
The quote are required! This is due to the forwarding of parameters to a svl script via a process, which unfortunately requires a very specific format. It is a non-trivial task to fix this dynamically in python. Thank the svl developers for this one.
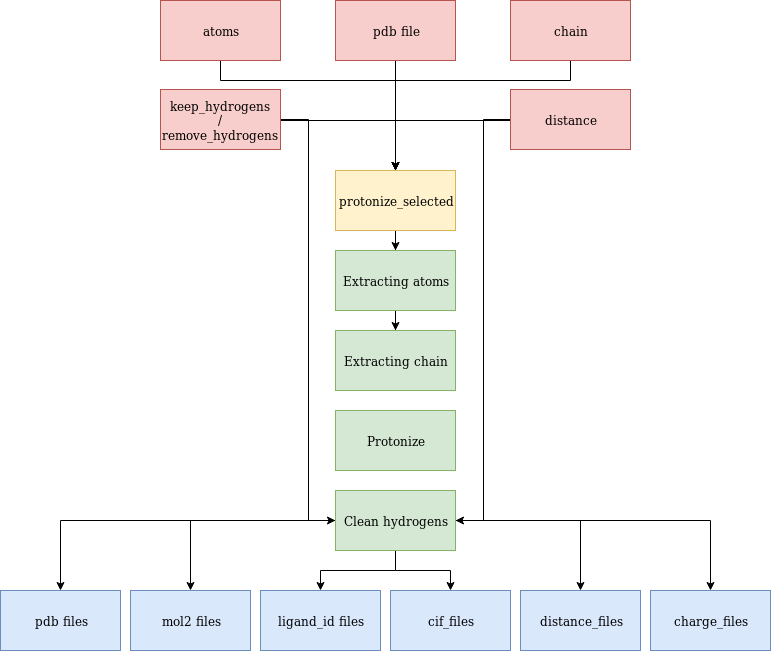
The generated results are very similar to the ones mentioned for protonize
promoe clean-hydrogens¶
Help menu:
____ ____ ___ __ __ ___ _____
| _ \| _ \ / _ \| \/ |/ _ \| ____|
| |_) | |_) | | | | |\/| | | | | _|
| __/| _ <| |_| | | | | |_| | |___
|_| |_| \_ \___/|_| |_|\___/|_____|
Usage: promoe clean-hydrogens [OPTIONS]
- Options:
--mol2 TEXT Path to the mol2 file[required] --distance INTEGER The maximal distance of the ligand to the surrounding water atoms after protonization, that should be kept. Default is 4. Hence, all water atoms within distance 4 to the ligand are not removed! --help Show this message and exit.

clean-hydrogens only generates distance files and the modified mol2 file.